import pandas as pd
import numpy as np
import seaborn as sns
import matplotlib.pyplot as plt
# 통계 검정
from scipy.stats import f_oneway
from scipy.stats import chi2_contingency
# ml
from sklearn.model_selection import train_test_split
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import classification_report, confusion_matrix, roc_auc_score
from sklearn.preprocessing import StandardScaler, LabelEncoder
from sklearn.pipeline import Pipeline
from sklearn.impute import SimpleImputer
# 모델 후보
from sklearn.linear_model import LogisticRegression
from sklearn.svm import SVC
# pytorch
import torch
import torch.nn as nn
import torch.optim as optim
from torch.utils.data import Dataset, DataLoader, random_split
from sklearn.preprocessing import StandardScaler
from sklearn.metrics import classification_report, confusion_matrix, ConfusionMatrixDisplaydata
| Column name | Original column name | Details |
|---|---|---|
| sample_id | Sample ID | Unique string identifying each subject |
| patient_cohort | Patient’s Cohort | Cohort 1: previously used samples; Cohort 2: newly added samples |
| sample_origin | Sample Origin | BPTB: Barts Pancreas Tissue Bank, London, UK; ESP: Spanish National Cancer Research Centre, Madrid, Spain; LIV: Liverpool University, UK; UCL: University College London, UK |
| age | Age | Age in years |
| sex | Sex | M = male, F = female |
| diagnosis | Diagnosis (1=Control, 2=Benign, 3=PDAC) | 1 = control (no pancreatic disease), 2 = benign hepatobiliary disease (119 of which are chronic pancreatitis), 3 = Pancreatic ductal adenocarcinoma (pancreatic cancer) |
| stage | Stage | For those with pancreatic cancer, one of IA, IB, IIA, IIIB, III, IV |
| benign_sample_diagnosis | Benign Samples Diagnosis | For those with a benign, non-cancerous diagnosis, what was the diagnosis? |
| plasma_CA19_9 | Plasma CA19-9 U/ml | Blood plasma levels of CA 19–9, often elevated in pancreatic cancer. Only assessed in 350 patients. |
| creatinine | Creatinine mg/ml | Urinary biomarker of kidney function |
| LYVE1 | LYVE1 ng/ml | Urinary levels of Lymphatic vessel endothelial hyaluronan receptor 1, may play a role in tumor metastasis |
| REG1B | REG1B ng/ml | Urinary levels of a protein that may be associated with pancreas regeneration |
| TFF1 | TFF1 ng/ml | Urinary levels of Trefoil Factor 1, related to regeneration and repair of urinary tract |
| REG1A | REG1A ng/ml | Urinary levels of a protein associated with pancreas regeneration. Only assessed in 306 patients. |
Data
df = pd.read_csv('../../../delete/Debernardi et al 2020 data.csv')df| sample_id | patient_cohort | sample_origin | age | sex | diagnosis | stage | benign_sample_diagnosis | plasma_CA19_9 | creatinine | LYVE1 | REG1B | TFF1 | REG1A | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | S1 | Cohort1 | BPTB | 33 | F | 1 | NaN | NaN | 11.7 | 1.83222 | 0.893219 | 52.948840 | 654.282174 | 1262.000 |
| 1 | S10 | Cohort1 | BPTB | 81 | F | 1 | NaN | NaN | NaN | 0.97266 | 2.037585 | 94.467030 | 209.488250 | 228.407 |
| 2 | S100 | Cohort2 | BPTB | 51 | M | 1 | NaN | NaN | 7.0 | 0.78039 | 0.145589 | 102.366000 | 461.141000 | NaN |
| 3 | S101 | Cohort2 | BPTB | 61 | M | 1 | NaN | NaN | 8.0 | 0.70122 | 0.002805 | 60.579000 | 142.950000 | NaN |
| 4 | S102 | Cohort2 | BPTB | 62 | M | 1 | NaN | NaN | 9.0 | 0.21489 | 0.000860 | 65.540000 | 41.088000 | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 585 | S549 | Cohort2 | BPTB | 68 | M | 3 | IV | NaN | NaN | 0.52026 | 7.058209 | 156.241000 | 525.178000 | NaN |
| 586 | S558 | Cohort2 | BPTB | 71 | F | 3 | IV | NaN | NaN | 0.85956 | 8.341207 | 16.915000 | 245.947000 | NaN |
| 587 | S560 | Cohort2 | BPTB | 63 | M | 3 | IV | NaN | NaN | 1.36851 | 7.674707 | 289.701000 | 537.286000 | NaN |
| 588 | S583 | Cohort2 | BPTB | 75 | F | 3 | IV | NaN | NaN | 1.33458 | 8.206777 | 205.930000 | 722.523000 | NaN |
| 589 | S590 | Cohort1 | BPTB | 74 | M | 3 | IV | NaN | 1488.0 | 1.50423 | 8.200958 | 411.938275 | 2021.321078 | 13200.000 |
590 rows × 14 columns
df.describe()| age | diagnosis | plasma_CA19_9 | creatinine | LYVE1 | REG1B | TFF1 | REG1A | |
|---|---|---|---|---|---|---|---|---|
| count | 590.000000 | 590.000000 | 350.000000 | 590.000000 | 590.000000 | 590.000000 | 590.000000 | 306.000000 |
| mean | 59.079661 | 2.027119 | 654.002944 | 0.855383 | 3.063530 | 111.774090 | 597.868722 | 735.281222 |
| std | 13.109520 | 0.804873 | 2430.317642 | 0.639028 | 3.438796 | 196.267110 | 1010.477245 | 1477.247724 |
| min | 26.000000 | 1.000000 | 0.000000 | 0.056550 | 0.000129 | 0.001104 | 0.005293 | 0.000000 |
| 25% | 50.000000 | 1.000000 | 8.000000 | 0.373230 | 0.167179 | 10.757216 | 43.961000 | 80.692000 |
| 50% | 60.000000 | 2.000000 | 26.500000 | 0.723840 | 1.649862 | 34.303353 | 259.873974 | 208.538500 |
| 75% | 69.000000 | 3.000000 | 294.000000 | 1.139482 | 5.205037 | 122.741013 | 742.736000 | 649.000000 |
| max | 89.000000 | 3.000000 | 31000.000000 | 4.116840 | 23.890323 | 1403.897600 | 13344.300000 | 13200.000000 |
for col in df.select_dtypes(include=['object', 'category']).columns:
print(f"\n📊 {col}")
print(df[col].value_counts(dropna=False))
📊 sample_id
S1 1
S588 1
S302 1
S288 1
S497 1
..
S282 1
S321 1
S323 1
S363 1
S590 1
Name: sample_id, Length: 590, dtype: int64
📊 patient_cohort
Cohort1 332
Cohort2 258
Name: patient_cohort, dtype: int64
📊 sample_origin
BPTB 409
LIV 132
ESP 29
UCL 20
Name: sample_origin, dtype: int64
📊 sex
F 299
M 291
Name: sex, dtype: int64
📊 stage
NaN 391
III 76
IIB 68
IV 21
IB 12
IIA 11
II 7
IA 3
I 1
Name: stage, dtype: int64
📊 benign_sample_diagnosis
NaN 382
Pancreatitis 41
Pancreatitis (Chronic) 35
Gallstones 21
Pancreatitis (Alcohol-Chronic) 11
Cholecystitis 9
Serous cystadenoma - NOS 7
Abdominal Pain 6
Choledocholiathiasis 6
Pancreatitis (Alcohol-Chronic-Pseuodcyst) 4
Pancreatitis (Gallstone) 4
Pancreatitis (Pseudocyst) 4
Pancreatitis (Idiopathic) 4
Pancreatitis (Autoimmune) 3
Serous microcystic adenoma 3
Gallstones - Incidental 3
Cholecystitis (Chronic) 3
Premalignant lesions-Mucinous cystadenoma-NOS 3
Gallstones 2
Gallbladder polyps 2
Premalignant lesions-Adenoma-NOS 2
Pancreatitis (Chronic-Pseudocyst) 2
Cholecystitis (Chronic) Cholelithiasis 2
Premalignant lesions-Villous adenoma-NOS 2
Pancreatitis (Gallstone-Pseudocyst) 1
Pancreatitis (Gallstone-Alcohol-Pseudocyst) 1
Pancreato-jejunostomy Anastomoses Stricture 1
Premalignant lesions-Tubulovillous adenoma-NOS 1
Pancreatitis (Chronic) Choledocholithiasis 1
Premalignant lesions-Mucinous cystadenocarcinoma-noninvasive 1
Pancreatitis (Hereditary-Chronic) 1
Pancreatitis (Hypertriglyceridemia) 1
Pancreatitis (Idiopathic) 1
Premalignant lesions-Tubular adenoma-NOS 1
Pancreatitis (Abscess) 1
Pancreatitis (Chronic) (Later became PDAC) 1
Pancreatitis (Alcohol) 1
Biliary Stricture (Secondary to Stent) 1
Cholecystitis 1
Cholecystitis (Chronic) Cholesterolsis 1
Choledochal Cyst 1
Choledocholiathiasis 1
Cholelithiasis with adenomyomatous hyperplasia 1
Duodenal Stricture 1
Duodenitis 1
Gallbladder Porcelain 1
Gastritis 1
Gastritis and Reflux 1
Ill defined lesion in uncinate process 1
Ischaemic Common Bile Duct Stricture 1
Pancreatitis 1
Pancreatitis (Acute) 1
Simple benign liver cyst 1
Name: benign_sample_diagnosis, dtype: int64- crosstab between sex
ct = pd.crosstab(df['sex'], df['diagnosis_label'])
print(ct)diagnosis_label Benign Control PDAC
sex
F 101 115 83
M 107 68 116- Within-sex Proportions
print("\nWithin-sex proportions (%):")
print((ct.div(ct.sum(axis=1), axis=0) * 100).round(2))
Within-sex proportions (%):
diagnosis_label Benign Control PDAC
sex
F 33.78 38.46 27.76
M 36.77 23.37 39.86남성이 많이 췌장암 발생하는 경향
성별 다른지 chi-square test
chi2, p, dof, expected = chi2_contingency(ct)
print(f"Chi-square test p-value = {p:.4f}")Chi-square test p-value = 0.0001다름
- visualization
prop = ct.div(ct.sum(axis=1), axis=0)
prop.plot(kind='bar', stacked=True, figsize=(6,4))
plt.title('Diagnosis distribution by Sex')
plt.ylabel('Proportion')
plt.legend(title='Diagnosis')
plt.tight_layout()
plt.show()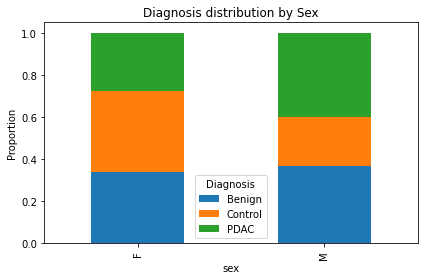
시각화해보면 딱 보임
diagnosis level visualization by Biomarker
df['diagnosis_label'] = df['diagnosis'].map({1:'Control', 2:'Benign', 3:'PDAC'})biomarkers = ['LYVE1', 'REG1B', 'TFF1', 'REG1A', 'plasma_CA19_9', 'creatinine']plt.figure(figsize=(14, 8))
for i, biomarker in enumerate(biomarkers, 1):
plt.subplot(2, 3, i)
sns.boxplot(data=df, x='diagnosis_label', y=biomarker, palette='Set2')
plt.title(biomarker)
plt.tight_layout()
plt.show()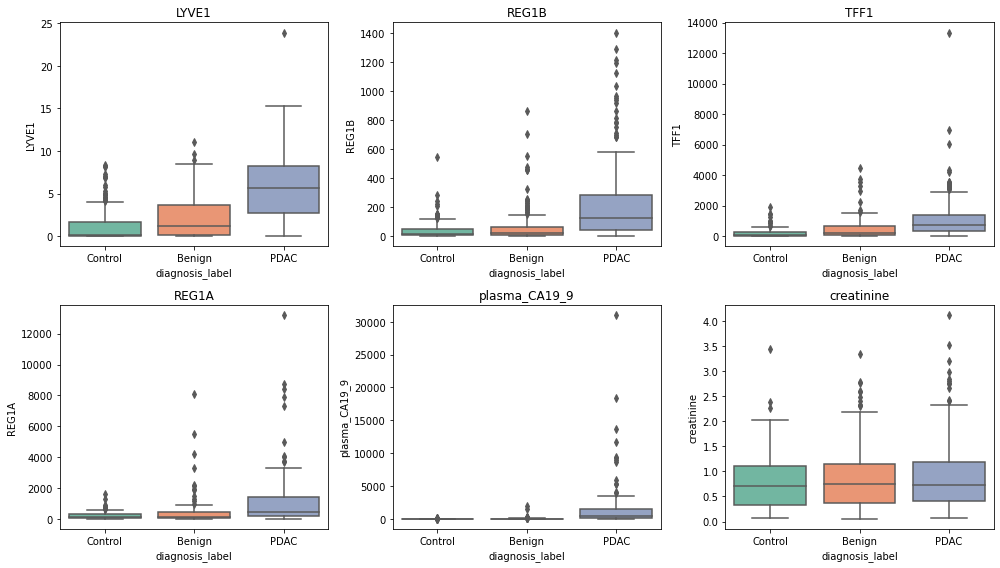
- 췌장암 환자 PDAC
- LYVE1이 높음. REG1B가 높음 TFF1이 높음, REF1A가 높음
Spearman Correlation
corr = df[biomarkers].corr(method="spearman", min_periods=20)
plt.figure(figsize=(8,7))
plt.imshow(corr, aspect="auto", interpolation="nearest")
plt.xticks(range(len(biomarkers)), biomarkers, rotation=45, ha="right")
plt.yticks(range(len(biomarkers)), biomarkers)
plt.title("Spearman correlation (biomarkers)")
plt.colorbar()
plt.tight_layout()
plt.show()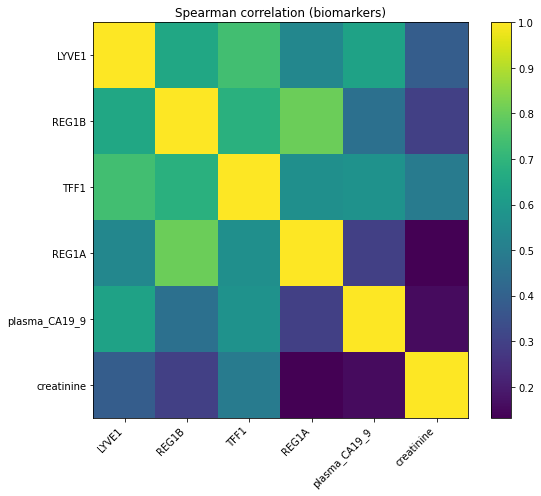
Statistical Test
for biomarker in biomarkers:
groups = [df[df['diagnosis_label'] == g][biomarker].dropna() for g in df['diagnosis_label'].unique()]
stat, p = f_oneway(*groups)
print(f"{biomarker}: p-value = {p:.4f}")LYVE1: p-value = 0.0000
REG1B: p-value = 0.0000
TFF1: p-value = 0.0000
REG1A: p-value = 0.0000
plasma_CA19_9: p-value = 0.0000
creatinine: p-value = 0.1894- dignose level 별로 그룹 간 차이가 있는지
- creatime 빼고 다 차이 있음
Random Forest
- target, features
X = df[biomarkers].fillna(0)
y = df['diagnosis']X_train, X_test, y_train, y_test = train_test_split(X, y, test_size=0.2, random_state=42)- randomforest
rf = RandomForestClassifier(random_state=42)
rf.fit(X_train, y_train)RandomForestClassifier(random_state=42)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
RandomForestClassifier(random_state=42)
- prediction
y_pred = rf.predict(X_test)
print(classification_report(y_test, y_pred)) precision recall f1-score support
1 0.76 0.76 0.76 41
2 0.61 0.59 0.60 39
3 0.85 0.87 0.86 38
accuracy 0.74 118
macro avg 0.74 0.74 0.74 118
weighted avg 0.74 0.74 0.74 118
Confusion Matrix
cm = confusion_matrix(y_test, y_pred, labels=[1,2,3])
plt.figure(figsize=(7,6))
plt.imshow(cm, interpolation="nearest")
plt.title("Confusion Matrix (RF)")
plt.xticks([0,1,2], ["1-Control","2-Benign","3-PDAC"], rotation=20)
plt.yticks([0,1,2], ["1-Control","2-Benign","3-PDAC"])
for i in range(3):
for j in range(3):
plt.text(j, i, cm[i, j], ha="center", va="center")
plt.colorbar()
plt.tight_layout()
plt.show()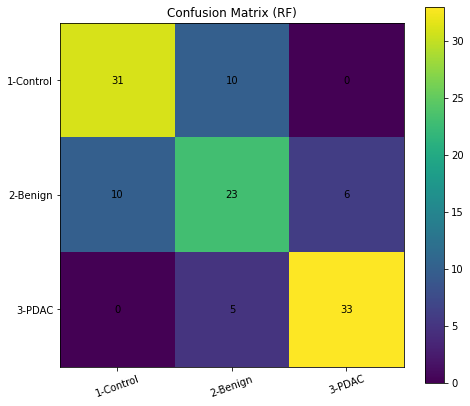
Check Feature Importance
importances = pd.Series(rf.feature_importances_, index=biomarkers).sort_values(ascending=False)
importancesplasma_CA19_9 0.241888
LYVE1 0.216604
TFF1 0.181022
REG1B 0.146198
creatinine 0.123882
REG1A 0.090405
dtype: float64sns.barplot(x=importances, y=importances.index)
plt.title('Feature Importance')
plt.show()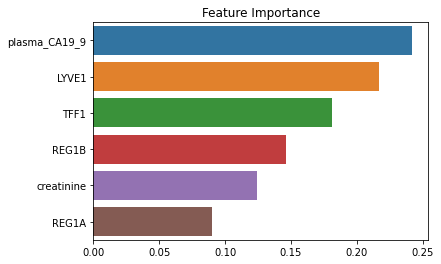
Logistic Regression
feature_cols = ["age", "creatinine", "LYVE1", "REG1B", "TFF1", "REG1A", "plasma_CA19_9"]
X = df[feature_cols].copy()
X["sex"] = df["sex"].map({"M":0, "F":1})
y = df["diagnosis"]X = X.fillna(X.median())
X_train, X_test, y_train, y_test = train_test_split(
X, y, test_size=0.25, random_state=42, stratify=y
)log_reg = Pipeline([
('scaler', StandardScaler()),
('clf', LogisticRegression(max_iter=200, multi_class='ovr'))
])
log_reg.fit(X_train, y_train)
print("\n--- Logistic Regression ---")
print(classification_report(y_test, log_reg.predict(X_test)))
--- Logistic Regression ---
precision recall f1-score support
1 0.62 0.63 0.62 46
2 0.60 0.52 0.56 52
3 0.75 0.84 0.79 50
accuracy 0.66 148
macro avg 0.66 0.66 0.66 148
weighted avg 0.66 0.66 0.66 148
svm = Pipeline([
('scaler', StandardScaler()),
('clf', SVC(kernel='rbf', probability=True))
])
svm.fit(X_train, y_train)
print("\n--- SVM (RBF kernel) ---")
print(classification_report(y_test, svm.predict(X_test)))
--- SVM (RBF kernel) ---
precision recall f1-score support
1 0.61 0.72 0.66 46
2 0.64 0.48 0.55 52
3 0.76 0.84 0.80 50
accuracy 0.68 148
macro avg 0.67 0.68 0.67 148
weighted avg 0.67 0.68 0.67 148
for name, model in [("LR", log_reg), ("SVM", svm)]:
y_proba = model.predict_proba(X_test)
auc = roc_auc_score(pd.get_dummies(y_test), y_proba, multi_class='ovr')
print(f"log_reg macro AUC: {auc:.3f}")log_reg macro AUC: 0.849
log_reg macro AUC: 0.843- AUC 별 차이 없음
biomarkers = ["age", "creatinine", "LYVE1", "REG1B", "TFF1", "REG1A", "plasma_CA19_9"]
X = df[biomarkers].copy()
X["sex"] = df["sex"].map({"M":0, "F":1})
y = df["diagnosis"] - 1 # 0,1,2 로 바꿈 (PyTorch용)X = X.fillna(X.median())
scaler = StandardScaler()
X_scaled = scaler.fit_transform(X)
X_train, X_test, y_train, y_test = train_test_split(X_scaled, y, test_size=0.25, random_state=42, stratify=y)
X_train = torch.tensor(X_train, dtype=torch.float32)
y_train = torch.tensor(y_train.values, dtype=torch.long)
X_test = torch.tensor(X_test, dtype=torch.float32)
y_test = torch.tensor(y_test.values, dtype=torch.long)- data loader
class BiomarkerDataset(Dataset):
def __init__(self, X, y):
self.X = X
self.y = y
def __len__(self):
return len(self.X)
def __getitem__(self, idx):
return self.X[idx], self.y[idx]train_dataset = BiomarkerDataset(X_train, y_train)
test_dataset = BiomarkerDataset(X_test, y_test)
train_loader = DataLoader(train_dataset, batch_size=16, shuffle=True)
test_loader = DataLoader(test_dataset, batch_size=16, shuffle=False)class MLPClassifier(nn.Module):
def __init__(self, input_dim, hidden_dims=[128, 64, 32], output_dim=3, dropout=0.4):
super().__init__()
self.layers = nn.Sequential(
nn.Linear(input_dim, hidden_dims[0]),
nn.ReLU(),
nn.Dropout(dropout),
nn.Linear(hidden_dims[0], hidden_dims[1]),
nn.ReLU(),
nn.Dropout(dropout),
nn.Linear(hidden_dims[1], hidden_dims[2]),
nn.ReLU(),
nn.Dropout(dropout),
nn.Linear(hidden_dims[2], output_dim)
)
def forward(self, x):
return self.layers(x)input_dim = X_train.shape[1]
model = MLPClassifier(input_dim=input_dim)
device = torch.device("cuda" if torch.cuda.is_available() else "cpu")
model = model.to(device)criterion = nn.CrossEntropyLoss()
optimizer = optim.Adam(model.parameters(), lr=0.001)
epochs = 100
train_losses, test_losses = [], []for epoch in range(epochs):
model.train()
total_loss = 0
for X_batch, y_batch in train_loader:
X_batch, y_batch = X_batch.to(device), y_batch.to(device)
optimizer.zero_grad()
output = model(X_batch)
loss = criterion(output, y_batch)
loss.backward()
optimizer.step()
total_loss += loss.item()
train_losses.append(total_loss / len(train_loader))
# Validation loss
model.eval()
val_loss = 0
with torch.no_grad():
for X_batch, y_batch in test_loader:
X_batch, y_batch = X_batch.to(device), y_batch.to(device)
output = model(X_batch)
val_loss += criterion(output, y_batch).item()
test_losses.append(val_loss / len(test_loader))
if epoch % 10 == 0:
print(f"Epoch [{epoch}/{epochs}] Train Loss: {train_losses[-1]:.4f} Val Loss: {test_losses[-1]:.4f}")Epoch [0/100] Train Loss: 1.0805 Val Loss: 1.0263
Epoch [10/100] Train Loss: 0.7961 Val Loss: 0.7018
Epoch [20/100] Train Loss: 0.7519 Val Loss: 0.6944
Epoch [30/100] Train Loss: 0.6993 Val Loss: 0.6584
Epoch [40/100] Train Loss: 0.7230 Val Loss: 0.6667
Epoch [50/100] Train Loss: 0.6858 Val Loss: 0.6535
Epoch [60/100] Train Loss: 0.6685 Val Loss: 0.6601
Epoch [70/100] Train Loss: 0.6517 Val Loss: 0.6542
Epoch [80/100] Train Loss: 0.6436 Val Loss: 0.6437
Epoch [90/100] Train Loss: 0.6599 Val Loss: 0.6618plt.plot(train_losses, label="Train Loss")
plt.plot(test_losses, label="Val Loss")
plt.legend()
plt.title("Training Curve")
plt.xlabel("Epoch")
plt.ylabel("Loss")
plt.show()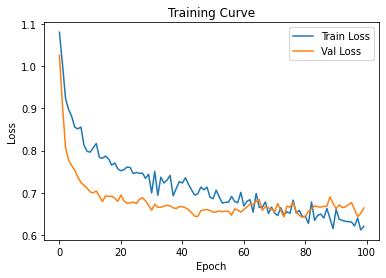
model.eval()MLPClassifier(
(layers): Sequential(
(0): Linear(in_features=8, out_features=128, bias=True)
(1): ReLU()
(2): Dropout(p=0.4, inplace=False)
(3): Linear(in_features=128, out_features=64, bias=True)
(4): ReLU()
(5): Dropout(p=0.4, inplace=False)
(6): Linear(in_features=64, out_features=32, bias=True)
(7): ReLU()
(8): Dropout(p=0.4, inplace=False)
(9): Linear(in_features=32, out_features=3, bias=True)
)
)with torch.no_grad():
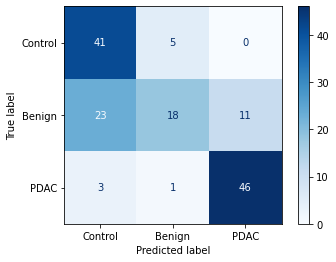
preds = model(X_test.to(device)).argmax(dim=1).cpu().numpy()print(classification_report(y_test, preds, target_names=["Control","Benign","PDAC"])) precision recall f1-score support
Control 0.61 0.89 0.73 46
Benign 0.75 0.35 0.47 52
PDAC 0.81 0.92 0.86 50
accuracy 0.71 148
macro avg 0.72 0.72 0.69 148
weighted avg 0.73 0.71 0.68 148
cm = confusion_matrix(y_test, preds)
disp = ConfusionMatrixDisplay(cm, display_labels=["Control","Benign","PDAC"])
disp.plot(cmap="Blues")
plt.show()